alvaMolecule is a software tool to visualise, analyse, curate and standardize your molecular dataset. alvaMolecule is free for academic and non-commercial use (Mauri, A., & Bertola, M. (2022). Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. International Journal of Molecular Sciences, 23(21), 12882. https://doi.org/10.3390/ijms232112882).
Molecular worksheet
alvaMolecule is conceived as a molecular worksheet where molecular datasets can be visualised both as a molecule grid or as a spreadsheet; additional data provided within SMILES and MDL files is automatically imported and can be used, together with the calculated descriptors and physicochemical properties, to sort and filter the molecular dataset.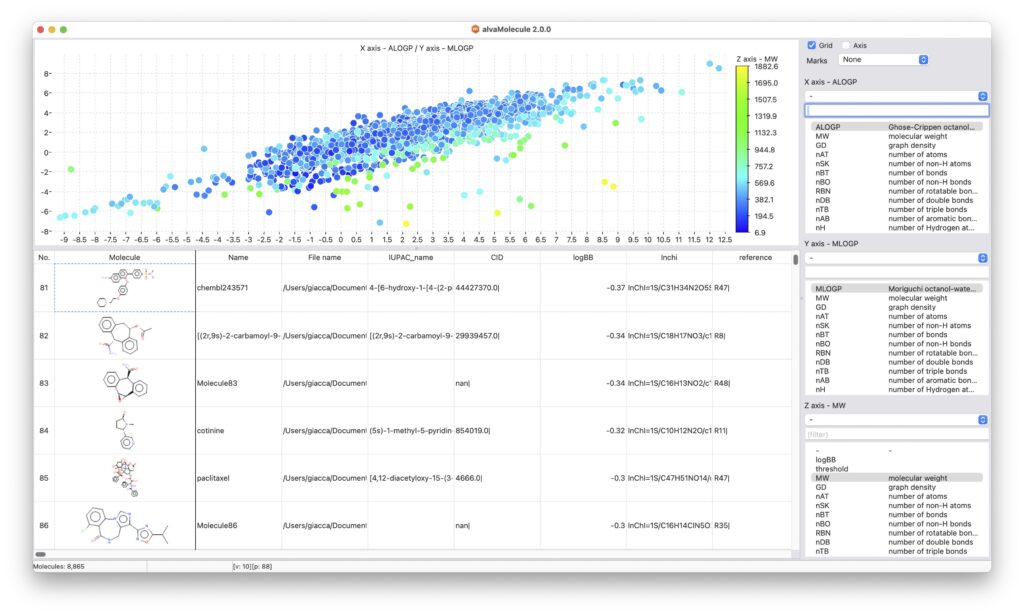
Molecule structure curation
alvaMolecule provides 10 predefined checkers that can help the identification of erroneous structure or for filtering specific structural features:
- Multiple structures
- Unusual valence atoms
- Covalent/Ionic bonds
- Total charge
- Isotopes
- Charged atoms
- No carbon atoms
- Non-standard atoms (H,B,C,N,O,P,S,F,Cl,Br,I)
- Aromaticity
- Radical atoms
Additionally, alvaMolecule provides the molecule structure verification using PubChem and GoogleChem services.
Molecule structure standardization
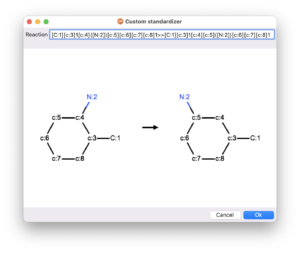 alvaMolecule provides 16 predefined standardizers that can be used to fix erroneous representation of molecules, to remove specific features from molecules or to standardize specific structural features:
alvaMolecule provides 16 predefined standardizers that can be used to fix erroneous representation of molecules, to remove specific features from molecules or to standardize specific structural features:
- Convert unusual covalent bonds to ionic bonds
- Add charge to quaternary nitrogens
- Remove exceeding hydrogens
- Add missing hydrogens
- Remove monoatomic fragments
- Retain biggest fragment
- Standardize nitro groups (-N(=O)=O)
- Standardize nitro groups (-[N+][O-]=O)
- Standardize azide groups
- Standardize diazo groups
- Clear isotopes
- Clear chirality
- Clear bond directions
- Remove radicals
- Neutraize atoms
- Neutralize molecules
In addition to the list of predefined standardizers, alvaMolecule allows the definition of custom standardizers. The Custom standardizer can be used to normalize molecular structure by defining a molecular transformation that can be applied on one reactant molecule. The transformation can be defined using the SMIRKS language.
Duplicates analysis
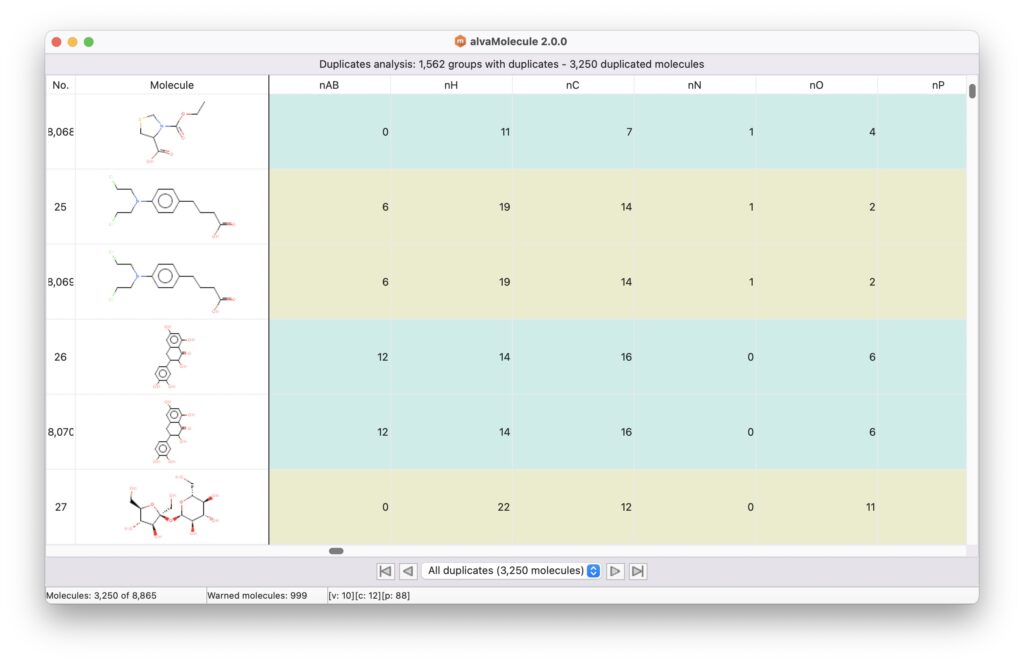 alvaMolecule can be used to identify duplicated structures or molecules having the same value for a specific column.
alvaMolecule can be used to identify duplicated structures or molecules having the same value for a specific column.
The duplicates analysis can be performed on molecular structures (Molecule) or on a column of the dataset:
- Molecule: the molecules having the same molecular structure are identified as duplicates. In this case, it is possible to select a set of features to be ignored during the duplicates analysis. For example, by selecting the Ignore stereochemistry option, the duplicates analysis will be performed ignoring the stereochemistry, i.e., molecules with the same molecular structures but with different stereochemistry will be considered as duplicates.
- Column: the molecules having the same value for the selected column are identified as duplicates.
Once the duplicates have been identified, they will be visualized in the main window. In addition, the number of identified duplicates is shown. The duplicates can be managed either manually (e.g., by deleting the duplicated molecules) or by using the automatic Manage duplicates option.
Scaffold analysis
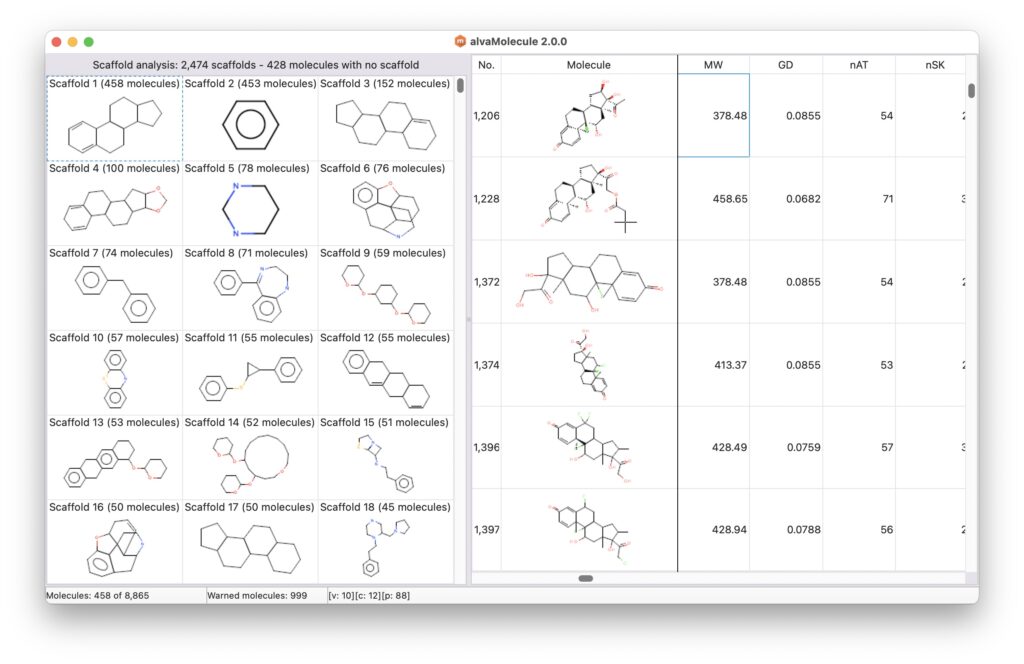 alvaMolecule can be used to identify the Bemis-Murcko frameworks (also known as scaffolds) of the loaded molecules (Bemis, G. W., & Murcko, M. A. (1996). The Properties of Known Drugs. 1. Molecular Frameworks. Journal of Medicinal Chemistry, 39(15), 2887–2893. https://doi.org/10.1021/jm9602928).
alvaMolecule can be used to identify the Bemis-Murcko frameworks (also known as scaffolds) of the loaded molecules (Bemis, G. W., & Murcko, M. A. (1996). The Properties of Known Drugs. 1. Molecular Frameworks. Journal of Medicinal Chemistry, 39(15), 2887–2893. https://doi.org/10.1021/jm9602928).
Once the scaffolds have been identified, they will be shown as a molecule grid in the main window. This molecule grid can be used to filter the molecules of the loaded dataset. The molecular scaffolds are ordered from the most frequent to the least one. If the dataset includes molecules with no scaffold, they are grouped together into the last group named No scaffold.
Edit, sort, filter and charting
alvaMolecule can be used to delete the undesired molecules in your dataset and to edit the automatically imported additional data.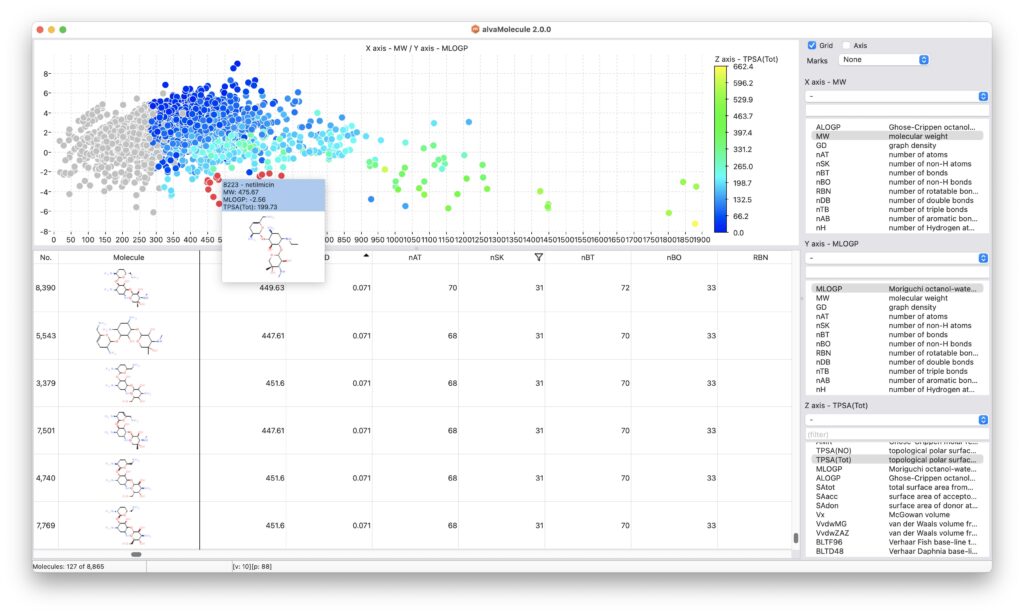
You can sort and filter molecules using:
- data loaded from molecule files
- calculated molecular descriptors
- calculated physicochemical properties
The same data can also be visualized and filtered using the charting tools provided within the software.
alvaMolecule can be used to filter molecules based on the presence or absence of a specific substructure defined using the SMARTS language.
Molecular descriptors and physicochemical properties
alvaMolecule calculates 88 molecular descriptors and physicochemical properties. Specifically alvaMolecule calculates a wide set of structural descriptors belonging to the constitutional and ring descriptors as implemented in alvaDesc.
Additionally, alvaMolecule calculates many physicochemical properties, drug-like and lead-like scores, among them several model-based physicochemical properties such as molar refractivity, topological polar surface area (TPSA), molecular volume estimations and two LogP models (Moriguchi and Ghose-Chippen octanol-water partition coefficient). A significant list of drug-like and lead-like scores is provided, including the well-known Lipinski alert index, which can be used to filter drug- and lead-like compounds.
Video
A short video introduction:
Platforms
The software is 64bit and it’s available for Windows, Linux and macOS.
Related tools
- alvaMolecule is the perfect tool to prepare your molecular dataset to be used by alvaDesc for the calculation of molecular descriptors and fingerprints and to check your molecules before applying QSAR models by alvaRunner.
- A tutorial showing how to build a QSAR model using Alvascience tools