
AlvaBuilder is a software tool for de novo molecular design (Mauri, A., & Bertola, M. (2024). AlvaBuilder: A Software for De Novo Molecular Design. Journal of Chemical Information and Modeling, 64(7), 2136–2142. https://doi.org/10.1021/acs.jcim.3c00610; Mauri, A., Bertola, M. (2022). De novo molecular design with genetic algorithms using alvaBuilder. ACS Fall 2022. https://doi.org/10.1021/scimeetings.2c00526). With its simple interface, it can be used to generate novel molecules having a desirable set of properties starting from a training set of your choice.
Genetic Algorithms
The drug-like chemical space has an estimated size of up to 1060 molecules. Searching through such a vast space can be a daunting activity. Genetic Algorithms can be used to tackle this problem by generating new molecules that have desired properties. Using alvaBuilder, new molecules can be designed to have certain molecular descriptor values, to have specified predictions for given QSAR / QSPR models or to be similar to target compounds. You can combine all the desired properties in the definition of a score function. The Genetic Algorithms of alvaBuilder will generate the new molecules by optimizing the defined score function.
Target molecular descriptors
A rule can be defined to generate molecules that have a target value for a specific molecular descriptor. The target can be defined as:
- Less than or equal to a given value
- Greater than or equal to a given value
- Equal to a given value
- Between two values (Range)
You can choose among a set of constitutional descriptors, such as the molecular weight (MW), the number of atoms (nAT) and bonds (nBT), the absolute and relative occurrence frequency of specific atom and bond types, as well as bonds and rings related properties like the rotatable bond number (RBN), the number of rings (nCIC), circuits (nCIR) and ring systems (NRS).
AlvaBuilder includes descriptors that can be used to evaluate specific structural features like the number of donor atoms for H-bonds (nHDon), the number of acceptor atoms for H-bonds (nHAcc) and the number of bridgehead (nBridgeHead) and spiro (nSpiro) atoms.
In addition to descriptors that can be used to define size and composition of a molecule, alvaBuilder provides a wide set of molecular properties, drug-like and lead-like indices that can be used to define the physicochemical characteristics of the newly designed molecules.
Molecular properties can be used to define a target value for molar refractivity (MRcons), octanol-water partition coefficient (LOGPcons), LogS aqueous solubility (ESOL), topological polar surface area (TPSA(tot), TPSA(NO)), surface area (SAtot, SAacc, SAdon) as well as McGowan (Vx) and van der Waals volumes (VvdwMG, VvdwZAZ).
Three base-line toxicities for Fish (BLTF96), Daphnia (BLTD48) and Algae (BLTA96) defined by Verhaar and based on Moriguchi LogP (MLOFP) are provided.
Since synthesizability is an important issue when dealing with de novo molecular generation, alvaBuilder includes also the synthetic accessibility score of drug-like molecules (SAscore) that provides a quantitative estimation of synthetic accessibility of molecules.
A diverse set of drug-like and lead-like indices are available, including the Lipinski’s rule of five (Ro5), LLS_01 that is based on the well known rule of three (Ro3) and the quantitative estimate of drug-likeness (QED).
Target QSAR/QSPR model
It is possible to define a rule to design molecules that yield a certain prediction value for a given regression or classification model. The QSAR / QSPR model must be contained in an alvaRunner project. The rule can also make use of the Applicability Domain (AD) i.e., rewarding those molecules that are inside the AD of the selected model and therefore preferring those compounds that fall within the theoretical region of the chemical space where a model’s prediction is considered to be reliable.
Target molecule
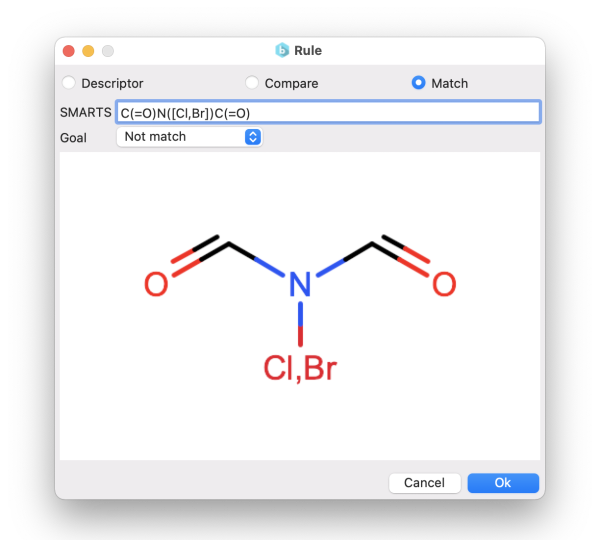
You can choose to generate compounds based on the similarity to target molecules. This can be achieved either by specifying:
- the whole molecule as a SMILES
- or through the SMARTS notation.
In the first case, the similarity is calculated using the Tanimoto distance on the molecular fingerprints. Instead, using the SMARTS, you can define molecular patterns that you want your final molecules to match or not to match (e.g., to avoid unwanted functional groups).
If needed, you can also define a list of molecular fragments that should always be included in each designed molecule.
Graphical user interface
An easy to use GUI allows you to manage the de novo molecule generation:
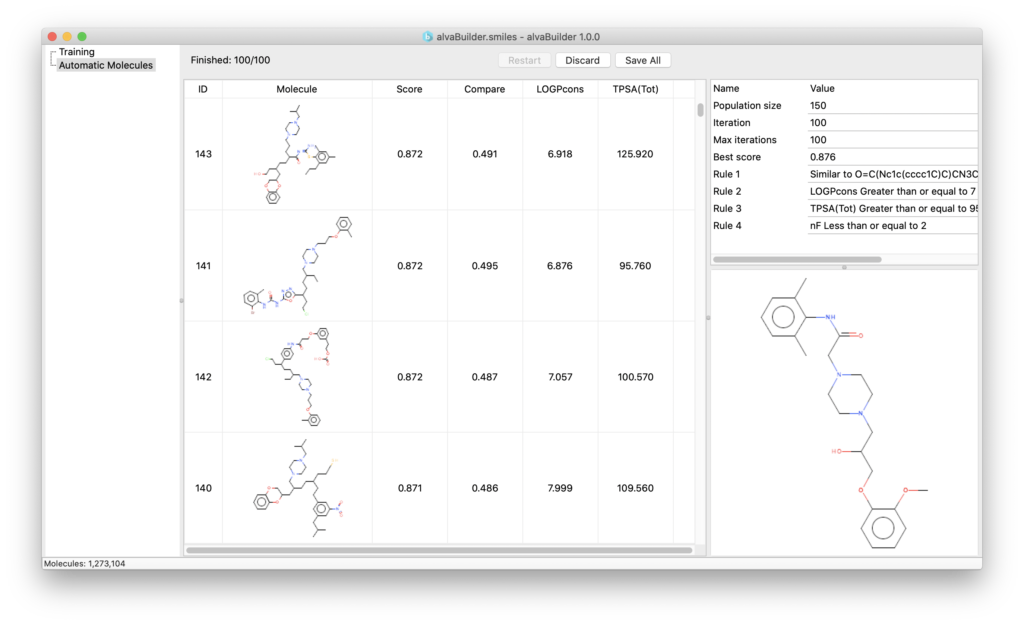
Using a simple step-by-step procedure (wizard) you can define the target properties you want the new molecules to have and start the generation.
Video
A short video introduction:
De novo molecular design with genetic algorithms using alvaBuilder:

Platforms
The software is 64bit and it’s available for Windows, Linux and macOS.