
alvaRunner is a software tool to apply Quantitative Structure Activity/Property Relationship (QSAR/QSPR) regression and classification models on a set of molecules. These models can be used to predict the biological, physicochemical and environmental properties of chemicals (Mauri, A., & Bertola, M. (2022). Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. International Journal of Molecular Sciences, 23(21), 12882. https://doi.org/10.3390/ijms232112882).
Apply the models
Without the need of any other software tool, alvaRunner calculates the descriptors and fingerprints needed in order to apply the given QSAR/QSPR regression and classification models.
The QSAR/QSPR models need to be created using alvaModel and they can be deployed, for example to allow other people:
- in the scientific community to reproduce your work (e.g., scientific paper)
- in you organisation to predict a certain property
User interface
alvaRunner is provided both as an easy to use command line tool and as an intuitive graphical interface. Using its interface, for every imported molecule, you can see the predicted targets and whether the molecule is inside or outside the defined model’s Applicability Domain and you can sort and filter any column by right-clicking the corresponding column header.
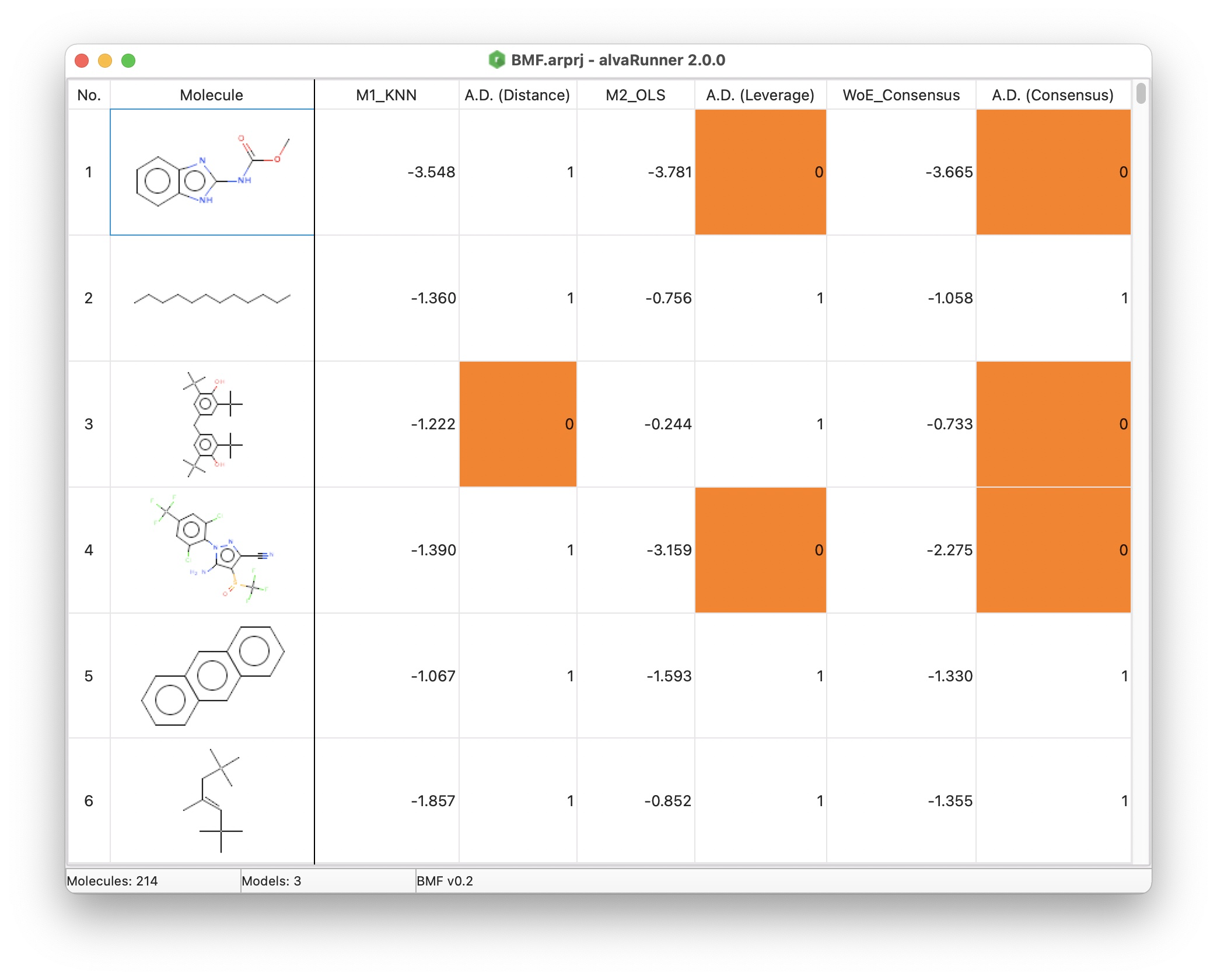
Prediction analysis
alvaRunner provides a functionality to evaluate a single prediction through a graphical interface called prediction detail.
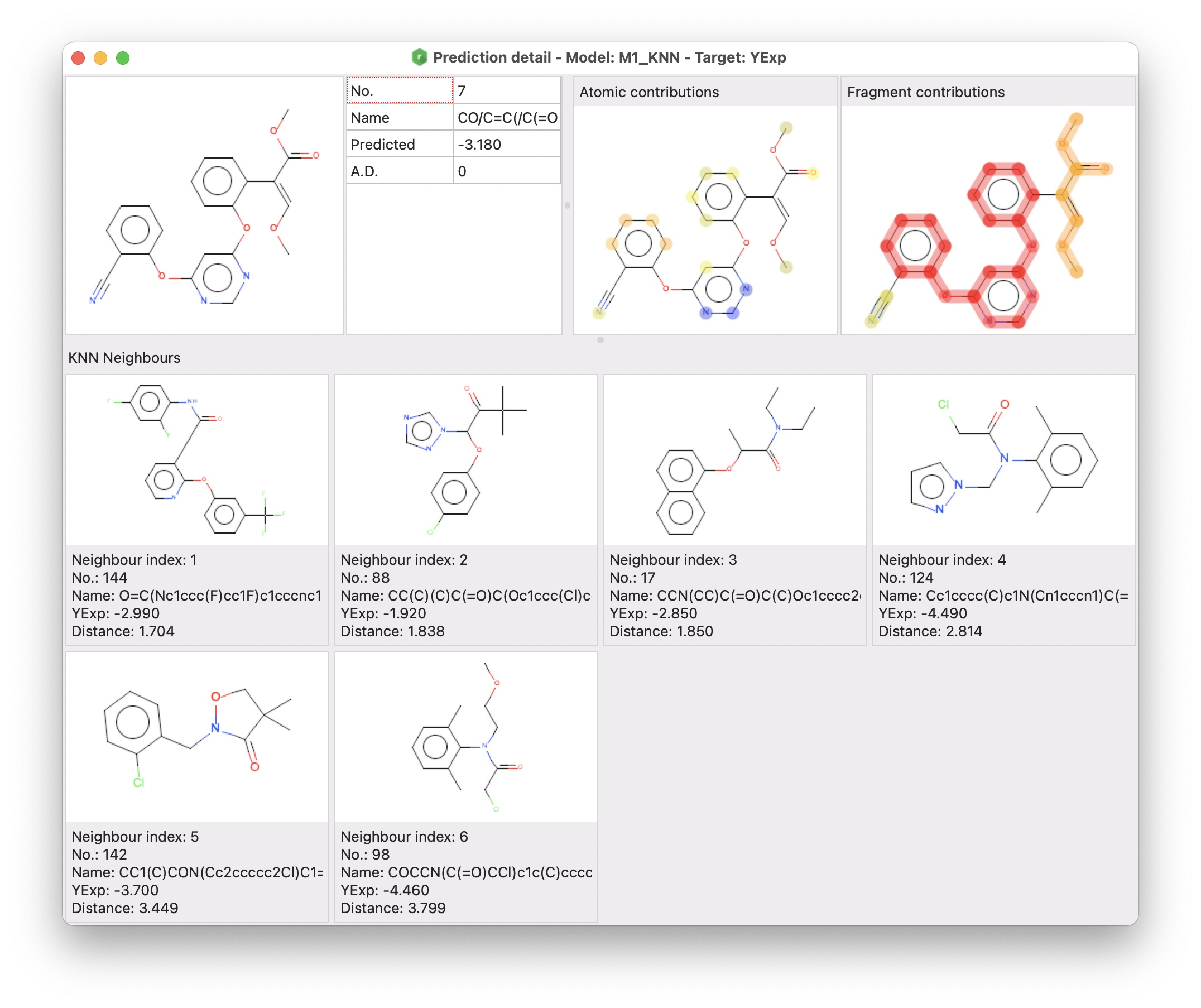
The prediction detail window includes three different sections:
- the target molecule and a grid including some information about the prediction
- atomic and fragment contributions (for all regression models except the consensus ones)
- KNN neighbours (for KNN models)
The atomic and fragment contributions are two visual representations of the contribution of atoms for the Atomic contributions, and framework and side chains for the Fragment contributions (Polishchuk, P. G., Kuz’min, V. E., Artemenko, A. G., & Muratov, E. N. (2013). Universal Approach for Structural Interpretation of QSAR/QSPR Models. Molecular Informatics, 32(9–10), 843–853. https://doi.org/10.1002/minf.201300029 – Riniker, S., & Landrum, G. A. (2013). Similarity maps – a visualization strategy for molecular fingerprints and machine-learning methods. Journal of Cheminformatics, 5(1), 43. https://doi.org/10.1186/1758-2946-5-43).
Video
A short video introduction:
Examples
A few example models were prepared to be applied to your molecules by using alvaRunner.
Platforms
The software is 64bit and it’s available for Windows, Linux and macOS. It is provided both as an easy to use command line tool and as an intuitive graphical interface.
Related tools
- alvaRunner can be integrated with KNIME using alvaRunner Plugin
- The descriptors and the fingerprints of the models are calculated using alvaDesc technology but without the need of having an alvaDesc license
- The models contained in the alvaRunner project must be prepared using alvaModel
- A tutorial showing how to build a QSAR model using Alvascience tools